Welcome to CHOC.org/health!

How to Explain Menstruation: Talking to Your Child About Periods
Struggling to teach your child about periods? Let the experts at CHOC help guide you through the conversation with these useful tips.

Window Safety
Every year, unintentional window falls send thousands of children ages 5 and younger to emergency departments nationwide. Learn how to protect your child.

The Facts About Difficult Eaters
It is tough to know how a child will react to foods being introduced into his or her diet. Learn the myths and facts about difficult eaters.

Side Effects of Chemotherapy and Radiation in Children
While chemotherapy and radiation are both treatments used to treat cancer, they differ in the way they work.

Flat Spots and the Shape of Your Baby’s Head
Babies who sleep on their back or in a car seat without changing positions for long periods of time can develop flat heads, a condition called plagiocephaly.

Kids and the Stomach Flu
Gastroenteritis, also known as the stomach flu, is inflammation in the digestive tract, including the stomach and the small and large intestines.
Precision Medicine and Pediatric Cancer Treatment
Precision medicine refers to the tailoring of medical treatment to the individual genetic characteristics of each patient’s disease.

The Facts about Type 1 Diabetes
Type 1 diabetes is an autoimmune disorder in which the body’s immune system damages the pancreas so that it can’t make enough insulin.

Suicide Prevention Among Teens and Children
What can I do if my kid has suicidal thoughts? Here’s everything you need to know about suicide prevention among teens and children.

Kids and Radiation Safety
Kids and radiation safety in imaging exams is a serious concern. Find out the important questions to ask your child's doctor or imaging facility.

Kids and Sleepwalking
Sleepwalking, also known as somnambulism, is a condition in which a sleeping person appears to be awake and exhibits behaviors associated with being awake, but is actually still asleep.
The Rewards of Giving Back
Community service can show kids and teens that giving their time, effort and kindness is more rewarding than receiving lots of presents.

High-Risk Pregnancy
Every family looks forward to a healthy pregnancy and to the birth of a healthy newborn. For some, there may be unexpected challenges along the way because an expectant mother has a high-risk pregnancy.
Understanding Pediatric Health Care
As a parent or guardian, you will make many decisions about your child’s health throughout their lifetime. Often, one of the first decisions you’ll make is what insurance to obtain.

New Car Seat Laws
New Car Seat Laws: What It Means for Your Family. Set to go into effect in 2017, children are required to rear-face until age 2. The new law does not apply to children who weigh more than 40 pounds or are 40 inches or taller.

Effects of Secondhand Smoke
Firsthand smoke comes from the toxins of cigarettes being inhaled directly. Secondhand smoke is breathed indirectly.
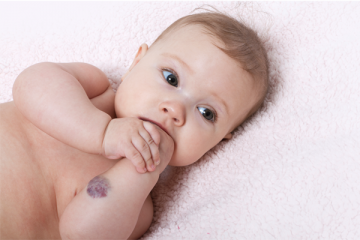
Vascular Anomalies
Vascular anomalies are congenital abnormalities of the vascular or lymphatic system. They are benign lesions that present anywhere on the body in the form of vascular birthmarks or masses.

Sprains and Strains
A sprain is a stretch or tear in a ligament, the bands of fibrous tissue that connect our bones at the joints. A strain is also a stretch or tear, but it affects the muscle itself or a tendon.

Identifying Epilepsy
Epilepsy is a seizure disorder characterized by having two or more “unprovoked” seizures. Epilepsy typically begins in infancy or early childhood.

The Truth About Genetics
Medical genetics involves the study of inherited diseases. The field includes genetic counseling and testing, and their application to patient care in the practice of medicine.
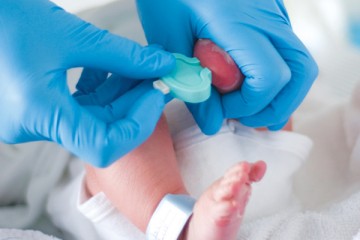
Metabolic Tests at Birth
Metabolic tests are done on newborns with very small blood samples that are taken at birth from every baby in California.

Having Asthma as an Athlete
Kids with asthma may experience these symptoms particularly during or after exercise. However, children with asthma who are well managed usually have very little difficulty with exercise.

Teens and Driving Safety
Even if you have a very responsible teen, the fact that they don’t have experience driving makes them a bigger risk. Driving is dangerous for all teens, and parents can require them to prove they are ready.

Dyslexia
Dyslexia is a fancy word for a developmental reading disability. It’s quite common and it has nothing to do with intelligence. There is a misperception that children with dyslexia see things backwards.

Helmets for Kids
Helmet for kids prevent moderate to more serious injuries, like bruising of the brain tissue itself. Helmets cushion the brain to prevent a more serious injuries.